
发布日期:2026-03-25 浏览次数:
在Nature上报道了一种分子编辑方法,能够将常见的醇官能团迁移至邻近位点,且其立体化学与区域化学结果均具有可预测性。该反应在激发态十钨酸盐聚阴离子催化的可逆氢原子转移条件下进行,通过一步1,2-酰氧基自由基迁移过程完成转化。文章链接DOI:10.1038/s47-4
醇羟基广泛存在于天然产物及药物分子中(图1a),但其在分子骨架上的分布往往集中于少数“热点”位置,限制了化学空间的多样性(图1b)。传统的羟基迁移多依赖酶催化,其高度电荷分离的过渡态需通过复杂的氢键网络稳定,小分子催化剂难以模拟。近年来,羰基迁移已有较多进展,但直接羟基迁移仍缺乏通用的小分子催化策略。
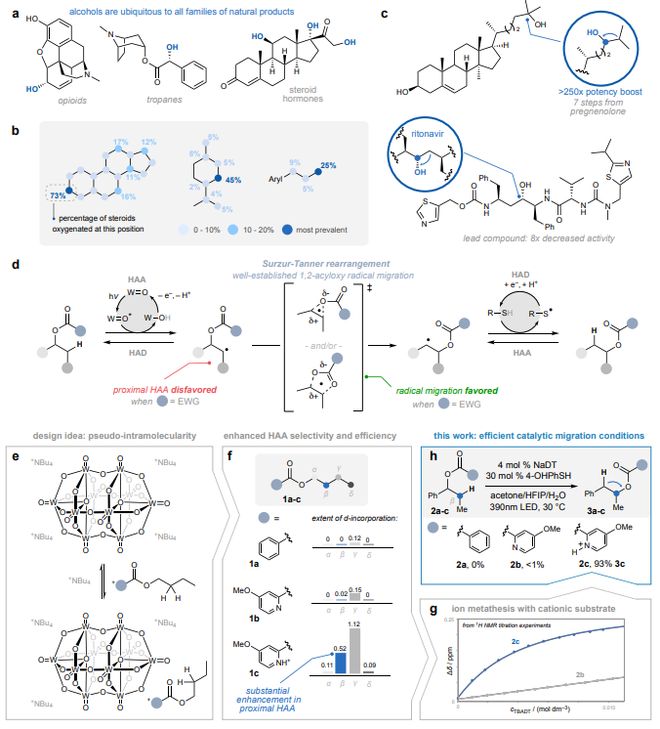
Alison E. Wendlandt教授课题组设想,通过激发态十钨酸盐(DT)聚阴离子促进的氢原子转移催化,利用合适的醇衍生酯的1,2-酰氧基自由基迁移,或称Surzur-Tanner重排,可以实现形式上的醇迁移(图1d)。这一核心机制特征对氢原子转移催化提出了电子需求上的根本性不匹配:促进1,2-自由基迁移所需的吸电子辅助基团,同时使邻位的C-H键对常见的亲电氢原子转移试剂(包括DT)的氢原子提取失活。
作者推断,在极性不匹配和位阻较大的位置形成自由基,可以通过向迁移性酯辅助基团中引入稳定的库仑相互作用、氢键或其他吸引性的非共价相互作用来增强。作者认为,对于能够与天然DT抗衡离子进行离子交换的阳离子酯基,可能会发生有效的、假分子内的氢原子提取(图1e)。如果成功,这种方法将重新调整氢原子提取和1,2-酰氧基自由基迁移步骤的内在电子需求,使得通过安装高度缺电子的阳离子酯辅助基团来加速这两个步骤成为可能。
为了验证这一假设,制备了一系列丁醇衍生物,其带有苯基、吡啶基和吡啶鎓酯基,并通过测量每个位点(α-δ)的H/D同位素交换的位置和程度,来评估自由基形成的位点和效率(图1f)。使用中性底物1a和1b时,氘的掺入几乎完全在γ-位点,这与缺电子酯基对β-位的诱导失活效应一致。而在吡啶鎓类似物1c的β-位检测到了显著的氘掺入水平,该类似物在基态下与四丁铵抗衡离子进行非选择性的离子交换(K = 1.4)(图1g)。除了增强的近端(β-位)自由基形成外,基于分子间竞争研究,阳离子底物1c的氢原子提取速率也快于中性底物1a。总的来说,这些设计要素使得2c到3c的自由基迁移得以高效进行,在最佳反应条件下(4 mol% NaDT,30 mol% 4-羟基苯硫酚,1.1当量 TMSOTf,丙酮/HFIP/H2O (8:1:1) 溶剂,390 nm LED照射,30 °C),20小时后NMR产率达到93%(图1h)。此处,TMSOTf作为三氟甲磺酸的原位前体,用于吡啶酯辅助基团的质子化。
从对映体富集的(R)-2b (98:2 e.r.) 出发,在标准30小时反应时间后,产物2c中观察到良好的对映体富集(93:7 e.r., 90% e.s.)。在较短的反应时间观察到更高的对映体特异性(如1小时后为95% e.s.),表明随时间推移出现的微小对映体纯度损失可能源于产物的缓慢背景外消旋化(图2a)。该反应在约1克规模上进行,对结果影响极小(80%分离产率,95:5 e.r.),在碱性条件下定量水解酯辅助基团,得到保持立体化学的游离醇(S)-4 (95:5 e.r.)。一系列具有给电子或吸电子芳环取代的衍生物(图2b)的反应结果揭示了在决速过渡态中正电荷的累积,与先前观察到的高度电荷分离的自由基迁移步骤的证据一致。
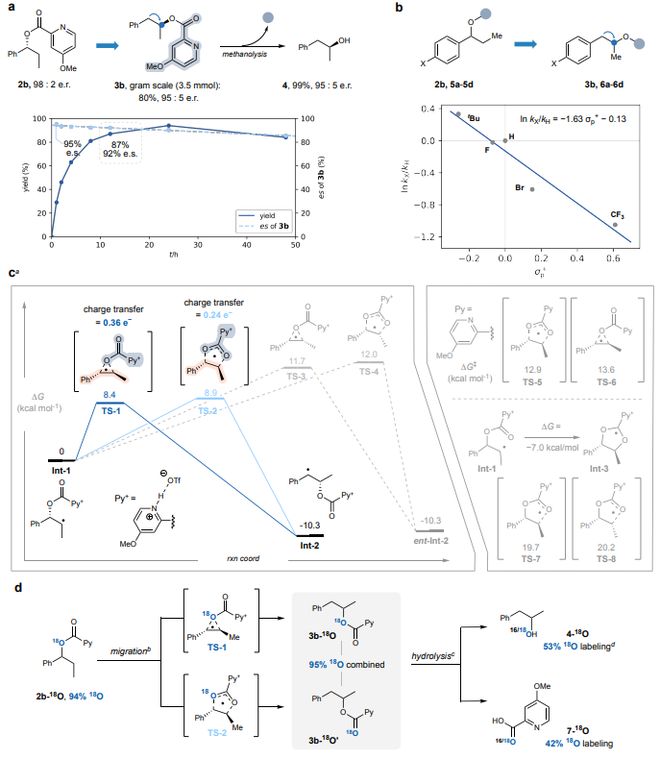
使用密度泛函理论对1,2-自由基迁移步骤进行的计算模拟,定位了两个能量几乎相等的过渡态结构,对应于协同的三元环(TS-1)和五元环(TS-2)自由基迁移过程(图2c)。在这两种几何结构中,苯基和甲基的反式排列(导致主要对映体Int-2的形成)在能量上优于相应的顺式构型结构,两种过渡态结构也显示出显著程度的电荷分离(分别为0.34 e和0.24 e),这与Hammett分析的结果一致(图2b)。
为了解析催化反应中三元环与五元环过渡态几何的贡献,将18O富集的底物探针2b-18O置于标准反应条件下,并在起始物料完全消耗后分离出产物3b-18O和3b-18O’(图2d)。酯水解后,测量了相应的吡啶甲酸(7-18O,42% 18O掺入)和醇(4-18O,53% 18O掺入)片段中的18O标记程度。在两个组分中均存在18O表明,在实验反应条件下,三元环和五元环(TS-1和TS-2)反应轨迹都对产物形成有贡献,这一发现与计算出的两条途径能量几乎相等的过渡态一致。
接下来,评估了一系列仲醇和叔醇衍生物作为C-O键迁移的底物(图3)。总的来说,成功的迁移发生在活化位置(如仲苄基或叔醇)向未活化位置(如仲脂肪族)的迁移。带有各种电子取代基的苄基仲醇衍生物都能顺利反应,形成相应的高苄醇产物6a-g(42-75%产率)。同样,线元环)也能反应,形成相应的邻位仲酯产物6j-p,且未检测到随后的产物仲酯向更远端位置的“过度迁移”。
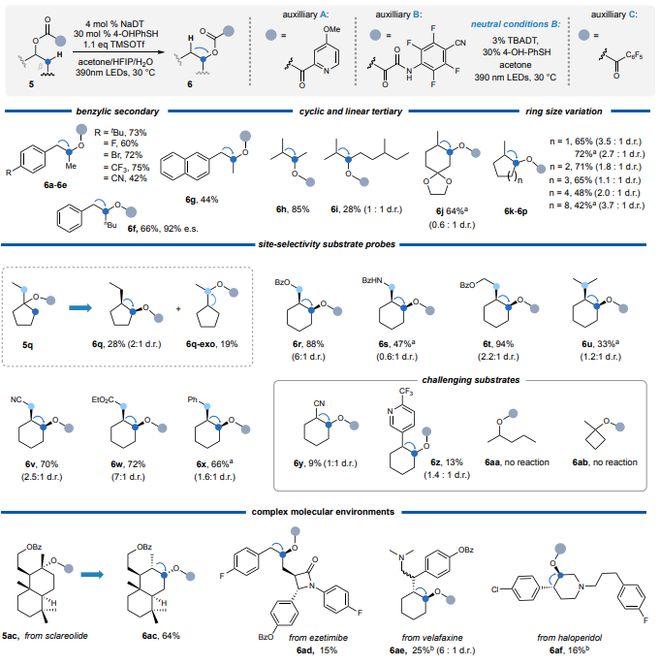
为了评估反应的位点选择性,制备了一系列具有多个潜在迁移位点的底物。例如,在1-乙基环戊醇衍生的酯5q的情况下,迁移可以发生在环外乙基亚甲基位置形成6q-exo,或者发生在环亚甲基PG电子官方网位置形成6q。在标准反应条件下,该底物反应生成两种产物,环戊醇产物6q的形成略有优势(比例为1.5:1)。迁移步骤的选择性可以通过存在电子去活化(如6r, 6s)、空间位阻(6u)或自由基稳定基团(6v, 6w, 6x)来调控。
在某些情况下,观察到形成了4-甲氧基吡啶甲酸,将其归因于背景布朗斯特酸催化的水解。针对这些情况,设计了一个中性草氨酸酯辅助基团和相应的中性反应条件,成功地阻止了不期望的水解。总体而言,精细调控迁移辅助基团的能力赋予了该方法显著的灵活性。在偕位带有吸电子基团的酯(如氰醇和杂芳基醇衍生的酯5y和5z)在标准反应条件下反应缓慢,而来自未活化仲醇的酯,如5aa,以及含有张力的环的5ab,在任何反应条件下均不反应。
为了测试该方法的官能团兼容性以及在合成后期阶段的可行性,从香紫苏内酯经3步制备的底物5ac,以65%的产率和良好的非对映选择性(14:1 顺/反)反应生成6ac。依泽替米贝衍生物的反应得到高苄基酯6ad,产率15%,并伴有非特异性分解产物。在文拉法辛和氟哌啶醇衍生的酯的情况下,碱性胺官能团的优先质子化需要使用2.0当量的HOTf(以TMSOTf形式),导致溶液中形成难溶的双阳离子物种。在探索了不同的迁移辅助基团后,确定了全氟苯甲酰酯是合适的底物,它们反应生成迁移产物6ae和6af,具有可用的产率和优异的反式选择性。
最后,作者探索了C-O键迁移与经典醇基安装方法相结合的战略意义(图4a)。例如,手性环氧化物官能团通过多样化的1,2-开环反应,为获得对映体富集的仲醇提供了无与伦比的途径。通过将邻位二取代产物与对映体特异性的醇基迁移相结合,可能获得形式上的“偕位”开环产物。将对映体纯的(S)-氧化苯乙烯8进行有机铜酸盐加成,形成仲醇9 (99:1 e.r.)。酯化,随后迁移(38%产率)和脱保护(84%产率),得到假对称的手性高苄醇10,具有良好的对映体富集度(96:4 e.r.,图4b)。使用现有的涉及对映面区分的方法PG电子官方网(如酮还原)则很难独立实现这类化合物的对映选择性合成。
接下来考察了从烯烃和酮开始的反应序列。通过中间体12实现烯烃11的形式上的1,3-水合以形成13(图4c)。酮同样可以通过与合适的有机金属亲核试剂(如格氏试剂或有机锂试剂)的1,2-加成,容易地转化为叔醇。初始叔醇的迁移允许通过形式上的1,2’-加成,将酮基用作β-官能化仲醇的前体。为了探索这种可能性,将胆甾烷-3-酮(14)与MeLi反应,形成非对映异构体15和16。15迁移后经水解,干净地得到醇17。相应的反式产物通过从16获得(图4d)。在这两种情况下,迁移都发生了完全的区域选择性,与类似甾体酮的烯醇化反应中报道的选择性一致。
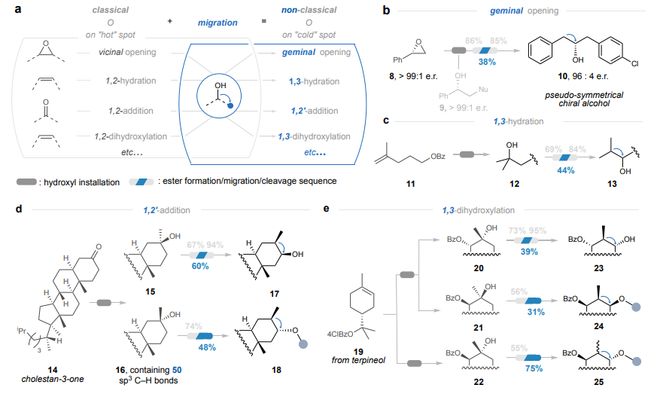
最后,虽然烯烃是1,2-二醇的理想合成子,但烯烃的1,3-二羟基化是一个长期存在的合成挑战。从O-4-氯苯甲酰基松油醇19开始,使用既定的二羟基化方案获得了顺式二醇衍生物20和21,以及反式二醇衍生物22。将20和21的酯置于迁移(和酯裂解)条件下,分别形成单一产物23和24,产率中等但物料平衡极佳。22的反应序列形成25,为两种非对映异构体的混合物(1:1 d.r.,图4e)。吡啶甲酸酯部分可以在存在其他苯甲酸酯的情况下,通过Cu(OAc)2催化的醇解选择性去除,且不会使带氧碳中心发生差向异构化。
Alison E. Wendlandt教授课题组报道了一种在氢原子转移光催化条件下,引入能够在底物和DT之间产生氢键和其他稳定非共价相互作用的酯辅助基团,通过1,2-酰氧基自由基迁移步骤实现醇衍生官能团迁移的策略。将该方法应用于合成后期,不仅能够实现醇官能团的精准重排,将其与常规的醇官能团引入方法相结合,更能为构建具有挑战性的氧化修饰模式提供全新的合成策略。
服务热线
